Risperdal: The following are discussed in more detail in Precautions: Increased mortality in elderly patients with dementia-related psychosis [see also Warnings]; Cerebrovascular adverse events, including stroke, in elderly patients with dementia-related psychosis; Neuroleptic malignant syndrome; Tardive dyskinesia; Metabolic Changes (Hyperglycemia and diabetes mellitus, Dyslipidemia, and Weight Gain); Hyperprolactinemia; Orthostatic hypotension; Falls; Leukopenia, neutropenia, and agranulocytosis; Potential for cognitive and motor impairment; Seizures; Dysphagia; Priapism; Disruption of body temperature regulation; Intraoperative Floppy Iris Syndrome (IFIS).
The most common adverse reactions in clinical trials (>5% and twice placebo) were parkinsonism, akathisia, dystonia, tremor, sedation, dizziness, anxiety, blurred vision, nausea, vomiting, upper abdominal pain, stomach discomfort, dyspepsia, diarrhea, salivary hypersecretion, constipation, dry mouth, increased appetite, increased weight, fatigue, rash, nasal congestion, upper respiratory tract infection, nasopharyngitis, and pharyngolaryngeal pain.
The most common adverse reactions that were associated with discontinuation from clinical trials (causing discontinuation in >1% of adults and/or >2% of pediatrics) were nausea, somnolence, sedation, vomiting, dizziness, and akathisia [see Clinical Trials Experience: Discontinuations Due to Adverse Reactions as follows].
The data described in this section are derived from a clinical trial database consisting of 9803 adult and pediatric patients exposed to one or more doses of RISPERDAL for the treatment of schizophrenia, bipolar mania, autistic disorder, and other psychiatric disorders in pediatrics and elderly patients with dementia. Of these 9803 patients, 2687 were patients who received RISPERDAL while participating in double-blind, placebo-controlled trials. The conditions and duration of treatment with RISPERDAL varied greatly and included (in overlapping categories) double-blind, fixed- and flexible-dose, placebo- or active-controlled studies and open-label phases of studies, inpatients and outpatients, and short-term (up to 12 weeks) and longer-term (up to 3 years) exposures. Safety was assessed by collecting adverse events and performing physical examinations, vital signs, body weights, laboratory analyses, and ECGs.
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Commonly-Observed Adverse Reactions in Double-Blind, Placebo-Controlled Clinical Trials - Schizophrenia: Adult Patients with Schizophrenia: Table 10 lists the adverse reactions reported in 2% or more of RISPERDAL-treated adult patients with schizophrenia in three 4- to 8-week, double-blind, placebo-controlled trials. (See Table 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pediatric Patients with Schizophrenia: Table 11 lists the adverse reactions reported in 5% or more of RISPERDAL-treated pediatric patients with schizophrenia in a 6-week double-blind, placebo-controlled trial. (See Table 11.)
Click on icon to see table/diagram/image
Commonly-Observed Adverse Reactions in Double-Blind, Placebo-Controlled Clinical Trials - Bipolar Mania: Adult Patients with Bipolar Mania: Table 12 lists the adverse reactions reported in 2% or more of RISPERDAL-treated adult patients with bipolar mania in four 3-week, double-blind, placebo-controlled monotherapy trials. (See Table 12.)
Click on icon to see table/diagram/image
Table 13 lists the adverse reactions reported in 2% or more of RISPERDAL-treated adult patients with bipolar mania in two 3-week, double-blind, placebo-controlled adjuvant therapy trials. (See Table 13.)
Click on icon to see table/diagram/image
Pediatric Patients with Bipolar Mania: Table 14 lists the adverse reactions reported in 5% or more of RISPERDAL-treated pediatric patients with bipolar mania in a 3-week double-blind, placebo-controlled trial. (See Table 14.)
Click on icon to see table/diagram/image
Commonly-Observed Adverse Reactions in Double-Blind, Placebo-Controlled Clinical Trials - Autistic Disorder: Table 15 lists the adverse reactions reported in 5% or more of RISPERDAL-treated pediatric patients treated for irritability associated with autistic disorder in two 8-week, double-blind, placebo-controlled trials and one 6-week double-blind, placebo-controlled study. (See Table 15.)
Click on icon to see table/diagram/image
Other Adverse Reactions Observed During the Clinical Trial Evaluation of Risperidone: The following additional adverse reactions occurred across all placebo-controlled, active-controlled, and open-label studies of RISPERDAL in adults and pediatric patients.
Blood and Lymphatic System Disorders: anemia, granulocytopenia, neutropenia.
Cardiac Disorders: sinus bradycardia, sinus tachycardia, atrioventricular block first degree, bundle branch block left, bundle branch block right, atrioventricular block.
Ear and Labyrinth Disorders: ear pain, tinnitus.
Endocrine Disorders: hyperprolactinemia.
Eye Disorders: ocular hyperemia, eye discharge, conjunctivitis, eye rolling, eyelid edema, eye swelling, eyelid margin crusting, dry eye, lacrimation increased, photophobia, glaucoma, visual acuity reduced.
Gastrointestinal Disorders: dysphagia, fecaloma, fecal incontinence, gastritis, lip swelling, cheilitis, aptyalism.
General Disorders: edema peripheral, thirst, gait disturbance, influenza-like illness, pitting edema, edema, chills, sluggishness, malaise, chest discomfort, face edema, discomfort, generalized edema, drug withdrawal syndrome, peripheral coldness, feeling abnormal.
Immune System Disorders: drug hypersensitivity.
Infections and Infestations: pneumonia, influenza, ear infection, viral infection, pharyngitis, tonsillitis, bronchitis, eye infection, localized infection, cystitis, cellulitis, otitis media, onychomycosis, acarodermatitis, bronchopneumonia, respiratory tract infection, tracheobronchitis, otitis media chronic.
Investigations: body temperature increased, blood prolactin increased, alanine aminotransferase increased, electrocardiogram abnormal, eosinophil count increased, white blood cell count decreased, blood glucose increased, hemoglobin decreased, hematocrit decreased, body temperature decreased, blood pressure decreased, transaminases increased.
Metabolism and Nutrition Disorders: decreased appetite, polydipsia, anorexia.
Musculoskeletal and Connective Tissue Disorders: joint stiffness, joint swelling, musculoskeletal chest pain, posture abnormal, myalgia, neck pain, muscular weakness, rhabdomyolysis.
Nervous System Disorders: balance disorder, disturbance in attention, dysarthria, unresponsive to stimuli, depressed level of consciousness, movement disorder, transient ischemic attack, coordination abnormal, cerebrovascular accident, speech disorder, syncope, loss of consciousness, hypoesthesia, tardive dyskinesia, dyskinesia, cerebral ischemia, cerebrovascular disorder, neuroleptic malignant syndrome, diabetic coma, head titubation.
Psychiatric Disorders: agitation, blunted affect, confusional state, middle insomnia, nervousness, sleep disorder, listlessness, libido decreased, and anorgasmia.
Renal and Urinary Disorders: enuresis, dysuria, pollakiuria, urinary incontinence.
Reproductive System and Breast Disorders: menstruation irregular, amenorrhea, gynecomastia, galactorrhea, vaginal discharge, menstrual disorder, erectile dysfunction, retrograde ejaculation, ejaculation disorder, sexual dysfunction, breast enlargement.
Respiratory, Thoracic, and Mediastinal Disorders: wheezing, pneumonia aspiration, sinus congestion, dysphonia, productive cough, pulmonary congestion, respiratory tract congestion, rales, respiratory disorder, hyperventilation, nasal edema.
Skin and Subcutaneous Tissue Disorders: erythema, skin discoloration, skin lesion, pruritus, skin disorder, rash erythematous, rash papular, rash generalized, rash maculopapular, acne, hyperkeratosis, seborrheic dermatitis.
Vascular Disorders: hypotension, flushing.
Additional Adverse Reactions Reported with RISPERDAL CONSTA: The following is a list of additional adverse reactions that have been reported during the premarketing evaluation of RISPERDAL CONSTA, regardless of frequency of occurrence: Cardiac Disorders: bradycardia.
Ear and Labyrinth Disorders: vertigo.
Eye Disorders: blepharospasm.
Gastrointestinal Disorders: toothache, tongue spasm.
General Disorders and Administration Site Conditions: pain.
Infections and Infestations: lower respiratory tract infection, infection, gastroenteritis, subcutaneous abscess.
Injury and Poisoning: fall.
Investigations: weight decreased, gamma-glutamyltransferase increased, hepatic enzyme increased.
Musculoskeletal, Connective Tissue, and Bone Disorders: buttock pain.
Nervous System Disorders: convulsion, paresthesia.
Psychiatric Disorders: depression.
Skin and Subcutaneous Tissue Disorders: eczema.
Vascular Disorders: hypertension.
Discontinuations Due to Adverse Reactions: Schizophrenia - Adults: Approximately 7% (39/564) of RISPERDAL-treated patients in double-blind, placebo-controlled trials discontinued treatment due to an adverse reaction, compared with 4% (10/225) who were receiving placebo. The adverse reactions associated with discontinuation in 2 or more RISPERDAL-treated patients were: See Table 16.
Click on icon to see table/diagram/image
Discontinuation for extrapyramidal symptoms (including Parkinsonism, akathisia, dystonia, and tardive dyskinesia) was 1% in placebo-treated patients, and 3.4% in active control-treated patients in a double-blind, placebo- and active-controlled trial.
Schizophrenia - Pediatrics: Approximately 7% (7/106), of RISPERDAL-treated patients discontinued treatment due to an adverse reaction in a double-blind, placebo-controlled trial, compared with 4% (2/54) placebo‑treated patients. The adverse reactions associated with discontinuation for at least one RISPERDAL-treated patient were dizziness (2%), somnolence (1%), sedation (1%), lethargy (1%), anxiety (1%), balance disorder (1%), hypotension (1%), and palpitation (1%).
Bipolar Mania - Adults: In double-blind, placebo-controlled trials with RISPERDAL as monotherapy, approximately 6% (25/448) of RISPERDAL-treated patients discontinued treatment due to an adverse event, compared with approximately 5% (19/424) of placebo-treated patients. The adverse reactions associated with discontinuation in RISPERDAL-treated patients were: See Table 17.
Click on icon to see table/diagram/image
Bipolar Mania - Pediatrics: In a double-blind, placebo-controlled trial 12% (13/111) of RISPERDAL-treated patients discontinued due to an adverse reaction, compared with 7% (4/58) of placebo-treated patients. The adverse reactions associated with discontinuation in more than one RISPERDAL-treated pediatric patient were nausea (3%), somnolence (2%), sedation (2%), and vomiting (2%).
Autistic Disorder - Pediatrics: In the two 8-week, placebo-controlled trials in pediatric patients treated for irritability associated with autistic disorder (n = 156), one RISPERDAL-treated patient discontinued due to an adverse reaction (Parkinsonism), and one placebo-treated patient discontinued due to an adverse event.
Dose Dependency of Adverse Reactions in Clinical Trials: Extrapyramidal Symptoms: Data from two fixed-dose trials in adults with schizophrenia provided evidence of dose-relatedness for extrapyramidal symptoms associated with RISPERDAL treatment.
Two methods were used to measure extrapyramidal symptoms (EPS) in an 8-week trial comparing 4 fixed doses of RISPERDAL (2, 6, 10, and 16 mg/day), including (1) a Parkinsonism score (mean change from baseline) from the Extrapyramidal Symptom Rating Scale, and (2) incidence of spontaneous complaints of EPS: See Table 18.
Click on icon to see table/diagram/image
Similar methods were used to measure extrapyramidal symptoms (EPS) in an 8-week trial comparing 5 fixed doses of RISPERDAL (1, 4, 8, 12, and 16 mg/day): See Table 19.
Click on icon to see table/diagram/image
Dystonia: Class Effect: Symptoms of dystonia, prolonged abnormal contractions of muscle groups, may occur in susceptible individuals during the first few days of treatment. Dystonic symptoms include: spasm of the neck muscles, sometimes progressing to tightness of the throat, swallowing difficulty, difficulty breathing, and/or protrusion of the tongue. While these symptoms can occur at low doses, they occur more frequently and with greater severity with high potency and at higher doses of first generation antipsychotic drugs. An elevated risk of acute dystonia is observed in males and younger age groups.
Other Adverse Reactions: Adverse event data elicited by a checklist for side effects from a large study comparing 5 fixed doses of RISPERDAL (1, 4, 8, 12, and 16 mg/day) were explored for dose-relatedness of adverse events. A Cochran-Armitage Test for trend in these data revealed a positive trend (p<0.05) for the following adverse reactions: somnolence, vision abnormal, dizziness, palpitations, weight increase, erectile dysfunction, ejaculation disorder, sexual function abnormal, fatigue, and skin discoloration.
Changes in Body Weight: Weight gain was observed in short-term, controlled trials and longer-term uncontrolled studies in adult and pediatric patients [see Precautions].
Changes in ECG Parameters: Between-group comparisons for pooled placebo-controlled trials in adults revealed no statistically significant differences between risperidone and placebo in mean changes from baseline in ECG parameters, including QT, QTc, and PR intervals, and heart rate. When all RISPERDAL doses were pooled from randomized controlled trials in several indications, there was a mean increase in heart rate of 1 beat per minute compared to no change for placebo patients. In short-term schizophrenia trials, higher doses of risperidone (8-16 mg/day) were associated with a higher mean increase in heart rate compared to placebo (4-6 beats per minute). In pooled placebo-controlled acute mania trials in adults, there were small decreases in mean heart rate, similar among all treatment groups.
In the two placebo-controlled trials in children and adolescents with autistic disorder (aged 5 ‑ 16 years) mean changes in heart rate were an increase of 8.4 beats per minute in the RISPERDAL groups and 6.5 beats per minute in the placebo group. There were no other notable ECG changes.
In a placebo-controlled acute mania trial in children and adolescents (aged 10 - 17 years), there were no significant changes in ECG parameters, other than the effect of RISPERDAL to transiently increase pulse rate (< 6 beats per minute). In two controlled schizophrenia trials in adolescents (aged 13 - 17 years), there were no clinically meaningful changes in ECG parameters including corrected QT intervals between treatment groups or within treatment groups over time.
Postmarketing Experience: The following adverse reactions have been identified during postapproval use of risperidone. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These adverse reactions include: alopecia, anaphylactic reaction, angioedema, atrial fibrillation, cardiopulmonary arrest, diabetic ketoacidosis in patients with impaired glucose metabolism, dysgeusia, floppy iris syndrome (intraoperative), hypoglycemia, hypothermia, ileus, inappropriate antidiuretic hormone secretion, intestinal obstruction, jaundice, mania, pancreatitis, pituitary adenoma, precocious puberty, pulmonary embolism, QT prolongation, sleep apnea syndrome, somnambulism, Stevens-Johnson syndrome and toxic epidermal necrolysis (SJS/TEN), sudden death, thrombocytopenia, thrombotic thrombocytopenic purpura, urinary retention, and water intoxication.
Risperdal Consta: The following are discussed in more detail in Precautions: Increased mortality in elderly patients with dementia-related psychosis [see also Warnings]; Cerebrovascular adverse events, including stroke, in elderly patients with dementia‑related psychosis; Neuroleptic malignant syndrome; Tardive dyskinesia; Metabolic changes; Hyperprolactinemia; Orthostatic hypotension; Falls; Leukopenia/Neutropenia and Agranulocytosis; Potential for cognitive and motor impairment; Seizures; Dysphagia; Priapism; Thrombotic Thrombocytopenic Purpura (TTP); Disruption of body temperature regulation; Avoidance of inadvertent injection into a blood vessel; Antiemetic effect; Increased sensitivity in patients with Parkinson's disease or those with dementia with Lewy bodies; Diseases or conditions that could affect metabolism or hemodynamic responses; Osteodystrophy and tumors in animals; Intraoperative Floppy Iris Syndrome (IFIS).
The most common adverse reactions in clinical trials in patients with schizophrenia (≥ 5%) were: headache, parkinsonism, dizziness, akathisia, fatigue, constipation, dyspepsia, sedation, weight increased, pain in extremity, and dry mouth. The most common adverse reactions in the double-blind, placebo-controlled periods of the bipolar disorder trials were weight increased (5% in the monotherapy trial) and tremor and parkinsonism (≥ 10% in the adjunctive treatment trial).
The most common adverse reactions that were associated with discontinuation from the 12‑week double-blind, placebo-controlled trial in patients with schizophrenia (causing discontinuation in ≥1% of patients) were agitation, depression, anxiety, and akathisia. Adverse reactions that were associated with discontinuation from the double-blind, placebo-controlled periods of the bipolar disorder trials were hyperglycemia (one patient in the monotherapy trial) and hypokinesia and tardive dyskinesia (one patient each in the adjunctive treatment trial).
The data described in this section are derived from a clinical trial database consisting of 2392 patients exposed to one or more doses of RISPERDAL CONSTA for the treatment of schizophrenia. Of these 2392 patients, 332 were patients who received RISPERDAL CONSTA while participating in a 12-week double-blind, placebo-controlled trial. Two hundred two (202) of the 332 were schizophrenia patients who received 25 mg or 50 mg RISPERDAL CONSTA. The conditions and duration of treatment with RISPERDAL CONSTA in the other clinical trials varied greatly and included (in overlapping categories) double-blind, fixed- and flexible-dose, placebo- or active-controlled studies and open-label phases of studies, inpatients and outpatients, and short-term (up to 12 weeks) and longer-term (up to 4 years) exposures. Safety was assessed by collecting adverse events and performing physical examinations, vital signs, body weights, laboratory analyses, and ECGs.
In addition to the studies in patients with schizophrenia, safety data are presented from a trial assessing the efficacy and safety of RISPERDAL CONSTA when administered as monotherapy for maintenance treatment in patients with bipolar I disorder. The subjects in this multi-center, double-blind, placebo-controlled study were adult patients who met DSM-IV criteria for Bipolar Disorder Type I and who were stable on risperidone (oral or long-acting injection), were stable on other antipsychotics or mood stabilizers, or were experiencing an acute episode. After a 3-week period of treatment with open-label oral risperidone (N=440), subjects who demonstrated an initial response to oral risperidone in this period and those who were stable on risperidone (oral or long-acting injection) at study entry entered into a 26-week stabilization period of open-label RISPERDAL CONSTA (N=501). Subjects who demonstrated a maintained response during this period were then randomized into a 24-month double-blind, placebo-controlled period in which they received RISPERDAL CONSTA (N=154) or placebo (N=149) as monotherapy. Subjects who relapsed or who completed the double-blind period could choose to enter an 8-week open-label RISPERDAL CONSTA extension period (N=160).
Safety data are also presented from a trial assessing the efficacy and safety of RISPERDAL CONSTA when administered as adjunctive maintenance treatment in patients with bipolar disorder. The subjects in this multi-center, double-blind, placebo-controlled study were adult patients who met DSM-IV criteria for Bipolar Disorder Type I or Type II and who experienced at least 4 episodes of mood disorder requiring psychiatric/clinical intervention in the previous 12 months, including at least 2 episodes in the 6 months prior to the start of the study. At the start of this study, all patients (N=275) entered into a 16-week open-label treatment phase in which they received RISPERDAL CONSTA in addition to continuing their treatment as usual, which consisted of various mood stabilizers (primarily lithium and valproate), antidepressants, and/or anxiolytics. Patients who reached remission at the end of this 16-week open-label treatment phase (N=139) were then randomized into a 52-week double-blind, placebo-controlled phase in which they received RISPERDAL CONSTA (N=72) or placebo (n=67) as adjunctive treatment in addition to continuing their treatment as usual. Patients who did not reach remission at the end of the 16-week open-label treatment phase could choose to continue to receive RISPERDAL CONSTA as adjunctive therapy in an open-label manner, in addition to continuing their treatment as usual, for up to an additional 36 weeks as clinically indicated for a total period of up to 52 weeks; these patients (N=70) were also included in the evaluation of safety.
Adverse events during exposure to study treatment were obtained by general inquiry and recorded by clinical investigators using their own terminology. Consequently, to provide a meaningful estimate of the proportion of individuals experiencing adverse events, events were grouped in standardized categories using MedDRA terminology.
Throughout this section, adverse reactions are reported. Adverse reactions are adverse events that were considered to be reasonably associated with the use of RISPERDAL CONSTA (adverse drug reactions) based on the comprehensive assessment of the available adverse event information. A causal association for RISPERDAL CONSTA often cannot be reliably established in individual cases. Further, because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The majority of all adverse reactions were mild to moderate in severity.
Commonly-Observed Adverse Reactions in Double-Blind, Placebo-Controlled Clinical Trials - Schizophrenia: Table 20 lists the adverse reactions reported in 2% or more of RISPERDAL CONSTA-treated patients with schizophrenia in one 12-week double-blind, placebo-controlled trial. (See Table 20.)
Click on icon to see table/diagram/image
Commonly-Observed Adverse Reactions in Double-Blind, Placebo-Controlled Clinical Trials - Bipolar Disorder: Table 21 lists the treatment-emergent adverse reactions reported in 2% or more of RISPERDAL CONSTA-treated patients in the 24-month double-blind, placebo-controlled treatment period of the trial assessing the efficacy and safety of RISPERDAL CONSTA when administered as monotherapy for maintenance treatment in patients with Bipolar I Disorder. (See Table 21.)
Click on icon to see table/diagram/image
Table 22 lists the treatment-emergent adverse reactions reported in 4% or more of patients in the 52-week double-blind, placebo-controlled treatment phase of a trial assessing the efficacy and safety of RISPERDAL CONSTA when administered as adjunctive maintenance treatment in patients with bipolar disorder. (See Table 22.)
Click on icon to see table/diagram/image
Other Adverse Reactions Observed During the Clinical Trial Evaluation of Risperidone: The following additional adverse reactions occurred in < 2% of the RISPERDAL CONSTA‑treated patients in the previously mentioned schizophrenia double-blind, placebo-controlled trial dataset, in < 2% of the RISPERDAL CONSTA-treated patients in the previously mentioned double-blind, placebo-controlled period of the monotherapy bipolar disorder trial dataset, or in < 4% of the RISPERDAL CONSTA-treated patients in the previously mentioned double-blind, placebo-controlled period of the adjunctive treatment bipolar disorder trial dataset. The following also includes additional adverse reactions reported at any frequency in RISPERDAL CONSTA-treated patients who participated in the open-label phases of the previously mentioned bipolar disorder studies and in other studies, including double-blind, active-controlled and open-label studies in schizophrenia and bipolar disorder.
Blood and lymphatic system disorders: anemia, neutropenia.
Cardiac disorders: tachycardia, atrioventricular block first degree, palpitations, sinus bradycardia, bundle branch block left, bradycardia, sinus tachycardia, bundle branch block right.
Ear and labyrinth disorders: ear pain, vertigo.
Endocrine disorders: hyperprolactinemia.
Eye disorders: conjunctivitis, visual acuity reduced.
Gastrointestinal disorders: diarrhea, vomiting, abdominal pain upper, abdominal pain, stomach discomfort, gastritis.
General disorders and administration site conditions: injection site pain, chest discomfort, chest pain, influenza like illness, sluggishness, malaise, induration, injection site induration, injection site swelling, injection site reaction, face edema.
Immune system disorders: hypersensitivity.
Infections and infestations: nasopharyngitis, influenza, bronchitis, urinary tract infection, rhinitis, respiratory tract infection, ear infection, pneumonia, lower respiratory tract infection, pharyngitis, sinusitis, viral infection, infection, localized infection, cystitis, gastroenteritis, subcutaneous abscess.
Injury and poisoning: fall, procedural pain.
Investigations: blood prolactin increased, alanine aminotransferase increased, electrocardiogram abnormal, gamma-glutamyl transferase increased, blood glucose increased, hepatic enzyme increased, aspartate aminotransferase increased, electrocardiogram QT prolonged, glucose urine present.
Metabolism and nutritional disorders: anorexia, hyperglycemia.
Musculoskeletal, connective tissue and bone disorders: posture abnormal, myalgia, back pain, buttock pain, muscular weakness, neck pain, musculoskeletal chest pain.
Nervous system disorders: coordination abnormal, dystonia, tardive dyskinesia, drooling, paresthesia, dizziness postural, convulsion, akinesia, hypokinesia, dysarthria.
Psychiatric disorders: insomnia, agitation, anxiety, sleep disorder, depression, initial insomnia, libido decreased, nervousness.
Renal and urinary disorders: urinary incontinence.
Reproductive system and breast disorders: galactorrhea, oligomenorrhea, erectile dysfunction, sexual dysfunction, ejaculation disorder, gynecomastia, breast discomfort, menstruation irregular, menstruation delayed, menstrual disorder, ejaculation delayed.
Respiratory, thoracic and mediastinal disorders: nasal congestion, pharyngolaryngeal pain, dyspnea, rhinorrhea.
Skin and subcutaneous tissue disorders: rash, eczema, pruritus generalized, pruritus.
Vascular disorders: hypotension, orthostatic hypotension.
Additional Adverse Reactions Reported with Oral RISPERDAL: The following is a list of additional adverse reactions that have been reported during the clinical trial evaluation of oral RISPERDAL, regardless of frequency of occurrence: Blood and Lymphatic Disorders: granulocytopenia.
Cardiac Disorders: atrioventricular block.
Ear and Labyrinth Disorders: tinnitus.
Eye Disorders: ocular hyperemia, eye discharge, eye rolling, eyelid edema, eye swelling, eyelid margin crusting, dry eye, lacrimation increased, photophobia, glaucoma.
Gastrointestinal Disorders: abdominal pain upper, dysphagia, fecaloma, abdominal discomfort, fecal incontinence, lip swelling, cheilitis, aptyalism.
General Disorders: thirst, feeling abnormal, gait disturbance, pitting edema, edema, chills, discomfort, generalized edema, drug withdrawal syndrome, peripheral coldness.
Immune System Disorders: drug hypersensitivity.
Infections and Infestations: tonsillitis, eye infection, cellulitis, otitis media, onychomycosis, acarodermatitis, bronchopneumonia, respiratory tract infection, tracheobronchitis, otitis media chronic.
Investigations: body temperature increased, heart rate increased, eosinophil count increased, white blood cell count decreased, hemoglobin decreased, blood creatine phosphokinase increased, hematocrit decreased, body temperature decreased, blood pressure decreased, transaminases increased.
Metabolism and Nutrition Disorders: polydipsia.
Musculoskeletal, Connective Tissue, and Bone Disorders: joint swelling, joint stiffness, rhabdomyolysis, torticollis.
Nervous System Disorders: hypertonia, balance disorder, dysarthria, unresponsive to stimuli, depressed level of consciousness, movement disorder, hypokinesia, parkinsonian rest tremor, transient ischemic attack, cerebrovascular accident, masked facies, speech disorder, loss of consciousness, muscle contractions involuntary, akinesia, cerebral ischemia, cerebrovascular disorder, neuroleptic malignant syndrome, diabetic coma, head titubation.
Psychiatric Disorders: blunted affect, confusional state, middle insomnia, listlessness, anorgasmia.
Renal and Urinary Disorders: enuresis, dysuria, pollakiuria.
Reproductive System and Breast Disorders: vaginal discharge, retrograde ejaculation, ejaculation disorder, ejaculation failure, breast enlargement.
Respiratory, Thoracic, and Mediastinal Disorders: epistaxis, wheezing, pneumonia aspiration, dysphonia, productive cough, pulmonary congestion, respiratory tract congestion, rales, respiratory disorder, hyperventilation, nasal edema.
Skin and Subcutaneous Tissue Disorders: erythema, skin discoloration, skin lesion, skin disorder, rash erythematous, rash papular, hyperkeratosis, dandruff, seborrheic dermatitis, rash generalized, rash maculopapular.
Vascular Disorders: flushing.
Discontinuations Due to Adverse Reactions: Schizophrenia: Approximately 11% (22/202) of RISPERDAL CONSTA-treated patients in the 12-week double-blind, placebo-controlled schizophrenia trial discontinued treatment due to an adverse event, compared with 13% (13/98) who received placebo. The adverse reactions associated with discontinuation in two or more RISPERDAL CONSTA-treated patients were: agitation (3%), depression (2%), anxiety (1%), and akathisia (1%).
Bipolar Disorder: In the 24-month double-blind, placebo-controlled treatment period of the trial assessing the efficacy and safety of RISPERDAL CONSTA when administered as monotherapy for maintenance treatment in patients with bipolar I disorder, 1 (0.6%) of 154 RISPERDAL CONSTA-treated patients discontinued due to an adverse reaction (hyperglycemia).
In the 52-week double-blind phase of the placebo-controlled trial in which RISPERDAL CONSTA was administered as adjunctive therapy to patients with bipolar disorder in addition to continuing with their treatment as usual, approximately 4% (3/72) of RISPERDAL CONSTA-treated patients discontinued treatment due to an adverse event, compared with 1.5% (1/67) of placebo-treated patients. Adverse reactions associated with discontinuation in RISPERDAL CONSTA-treated patients were: hypokinesia (one patient) and tardive dyskinesia (one patient).
Dose Dependency of Adverse Reactions in Clinical Trials: Extrapyramidal Symptoms: Two methods were used to measure extrapyramidal symptoms (EPS) in the 12-week double‑blind, placebo-controlled trial comparing three doses of RISPERDAL CONSTA (25 mg, 50 mg, and 75 mg) with placebo in patients with schizophrenia, including: (1) the incidence of spontaneous reports of EPS symptoms; and (2) the change from baseline to endpoint on the total score (sum of the subscale scores for parkinsonism, dystonia, and dyskinesia) of the Extrapyramidal Symptom Rating Scale (ESRS).
As shown in Table 3, the overall incidence of EPS-related adverse reactions (akathisia, dystonia, parkinsonism, and tremor) in patients treated with 25 mg RISPERDAL CONSTA was comparable to that of patients treated with placebo; the incidence of EPS-related adverse reactions was higher in patients treated with 50 mg RISPERDAL CONSTA.
The median change from baseline to endpoint in total ESRS score showed no worsening in patients treated with RISPERDAL CONSTA compared with patients treated with placebo: 0 (placebo group); -1 (25-mg group, significantly less than the placebo group); and 0 (50-mg group).
Dystonia: Class Effect: Symptoms of dystonia, prolonged abnormal contractions of muscle groups, may occur in susceptible individuals during the first few days of treatment. Dystonic symptoms include: spasm of the neck muscles, sometimes progressing to tightness of the throat, swallowing difficulty, difficulty breathing, and/or protrusion of the tongue. While these symptoms can occur at low doses, they occur more frequently and with greater severity with high potency and at higher doses of first generation antipsychotic drugs. An elevated risk of acute dystonia is observed in males and younger age groups.
Changes in ECG: The electrocardiograms of 202 schizophrenic patients treated with 25 mg or 50 mg RISPERDAL CONSTA and 98 schizophrenic patients treated with placebo in the 12-week double-blind, placebo-controlled trial were evaluated. Compared with placebo, there were no statistically significant differences in QTc intervals (using Fridericia's and linear correction factors) during treatment with RISPERDAL CONSTA.
The electrocardiograms of 227 patients with Bipolar I Disorder were evaluated in the 24-month double-blind, placebo-controlled period. There were no clinically relevant differences in QTc intervals (using Fridericia's and linear correction factors) during treatment with RISPERDAL CONSTA compared to placebo.
The electrocardiograms of 85 patients with bipolar disorder were evaluated in the 52-week double-blind, placebo-controlled trial. There were no statistically significant differences in QTc intervals (using Fridericia's and linear correction factors) during treatment with RISPERDAL CONSTA 25 mg, 37.5 mg, or 50 mg when administered as adjunctive treatment in addition to continuing treatment as usual compared to placebo.
Pain Assessment and Local Injection Site Reactions: The mean intensity of injection pain reported by patients with schizophrenia using a visual analog scale (0 = no pain to 100 = unbearably painful) decreased in all treatment groups from the first to the last injection (placebo: 16.7 to 12.6; 25 mg: 12.0 to 9.0; 50 mg: 18.2 to 11.8). After the sixth injection (Week 10), investigator ratings indicated that 1% of patients treated with 25 mg or 50 mg RISPERDAL CONSTA experienced redness, swelling, or induration at the injection site.
In a separate study to observe local-site tolerability in which RISPERDAL CONSTA was administered into the deltoid muscle every 2 weeks over a period of 8 weeks, no patient discontinued treatment due to local injection site pain or reaction. Clinician ratings indicated that only mild redness, swelling, or induration at the injection site was observed in subjects treated with 37.5 mg or 50 mg RISPERDAL CONSTA at 2 hours after deltoid injection. All ratings returned to baseline at the predose assessment of the next injection 2 weeks later. No moderate or severe reactions were observed in any subject.
Postmarketing Experience: The following adverse reactions have been identified during postapproval use of risperidone; because these reactions are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency: agranulocytosis, alopecia, anaphylactic reaction, angioedema, atrial fibrillation, blood cholesterol increased, blood triglycerides increased, diabetes mellitus, diabetic ketoacidosis in patients with impaired glucose metabolism, drug withdrawal syndrome neonatal, dysgeusia, floppy iris syndrome (intraoperative), hypoglycemia, hypothermia, ileus, inappropriate antidiuretic hormone secretion, intestinal obstruction, jaundice, mania, pancreatitis, priapism, QT prolongation, sleep apnea syndrome, somnambulism, Stevens-Johnson syndrome and toxic epidermal necrolysis (SJS/TEN), thrombocytopenia, urinary retention, and water intoxication. In addition, the following adverse reactions have been observed during postapproval use of RISPERDAL CONSTA: cerebrovascular disorders, including cerebrovascular accidents, and diabetes mellitus aggravated.
Retinal artery occlusion after injection of RISPERDAL CONSTA has been reported during postmarketing surveillance. This has been reported in the presence of abnormal arteriovenous anastomosis.
Serious injection site reactions including abscess, cellulitis, cyst, hematoma, necrosis, nodule, and ulcer have been reported with RISPERDAL CONSTA during postmarketing surveillance. Isolated cases required surgical intervention.
Very rarely, cases of anaphylactic reaction after injection with RISPERDAL CONSTA have been reported during postmarketing experience in patients who have previously tolerated oral risperidone.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image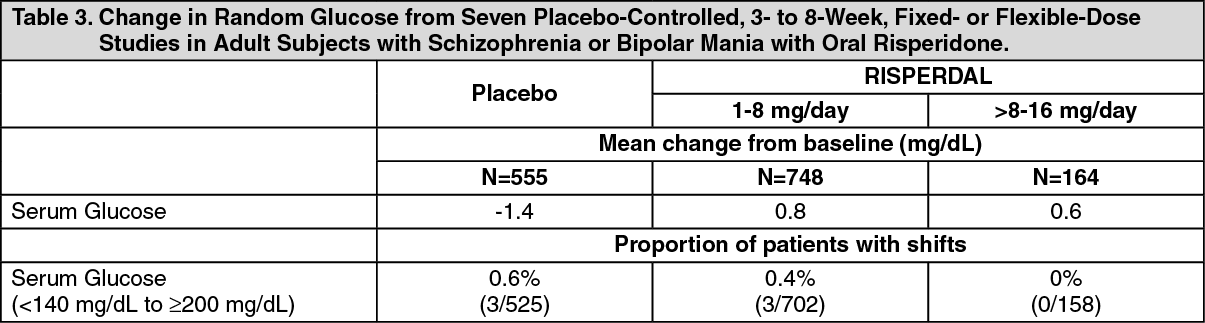 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image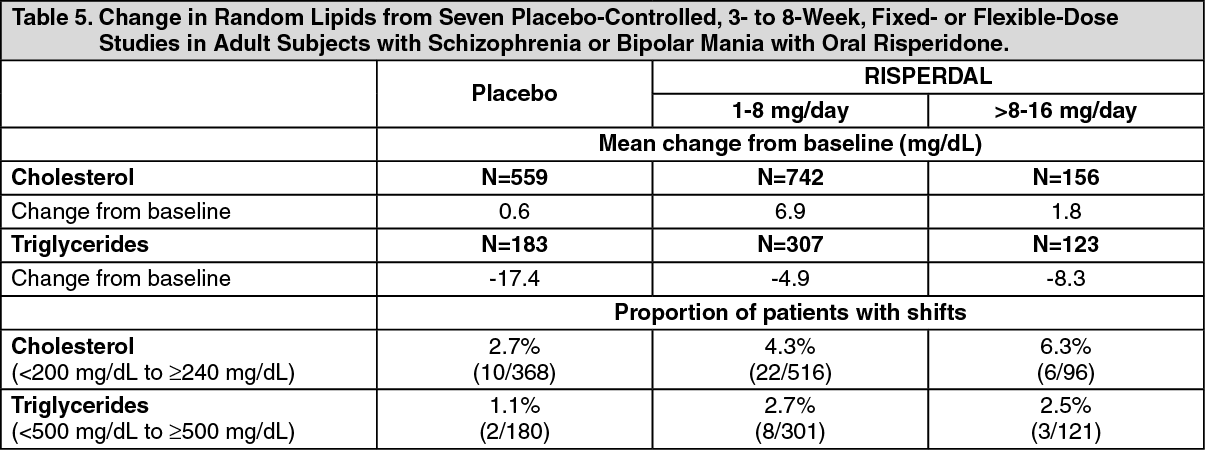 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image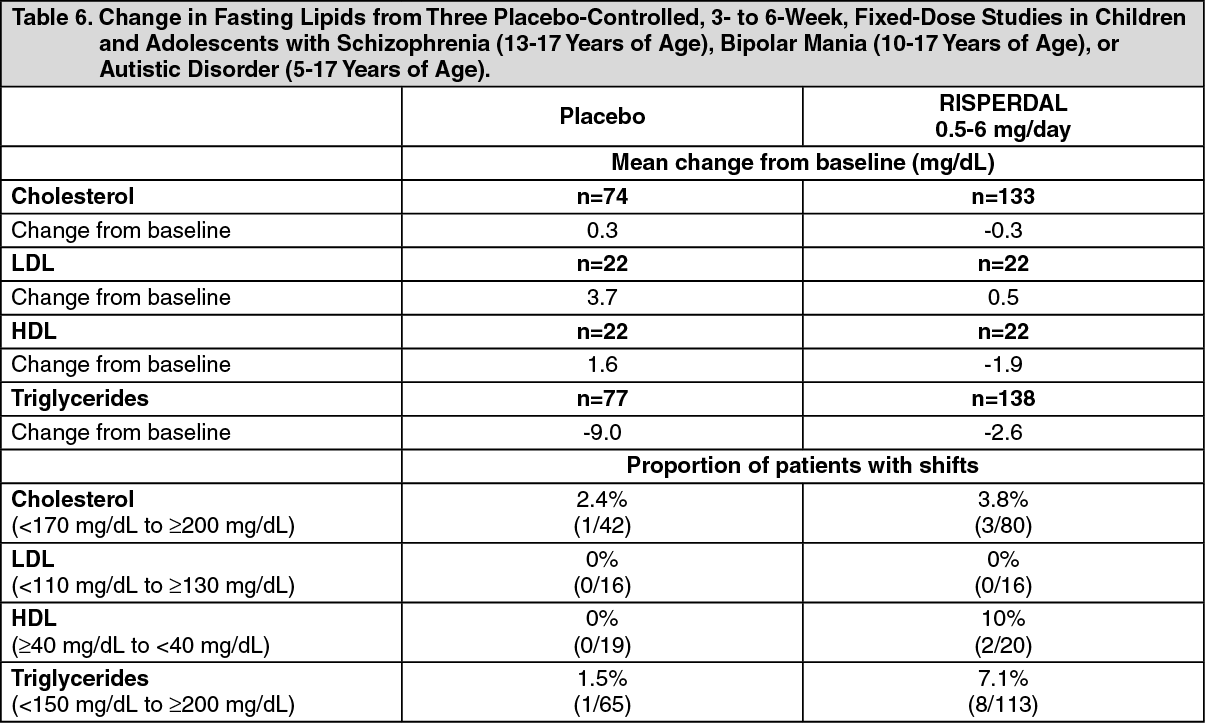 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image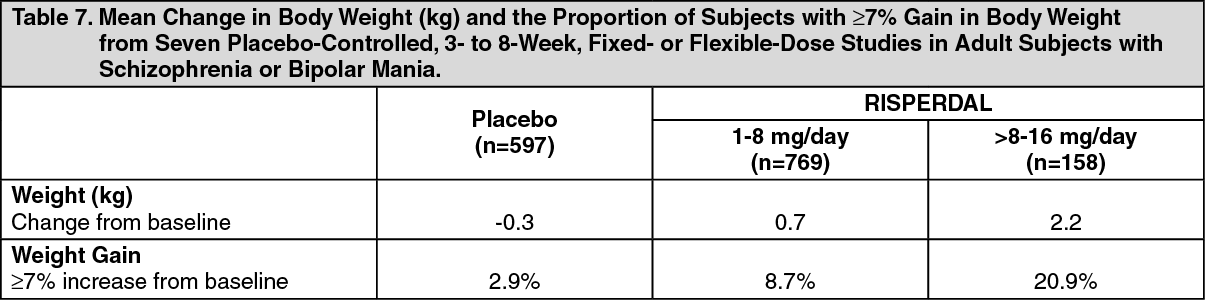 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image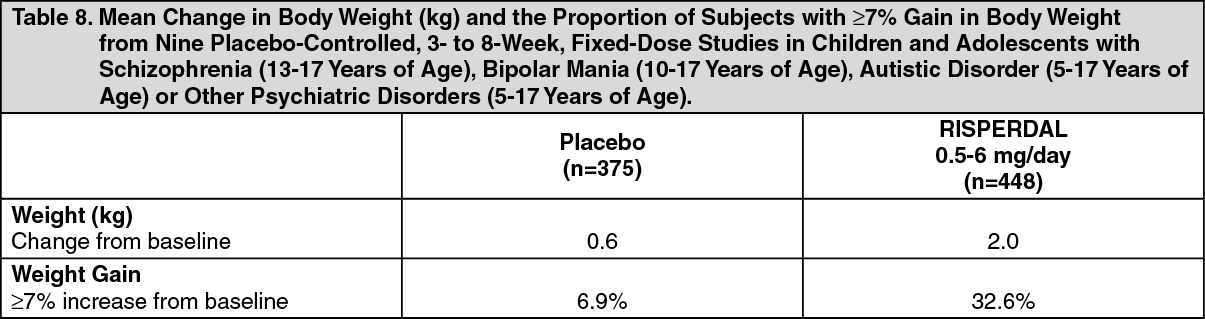 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image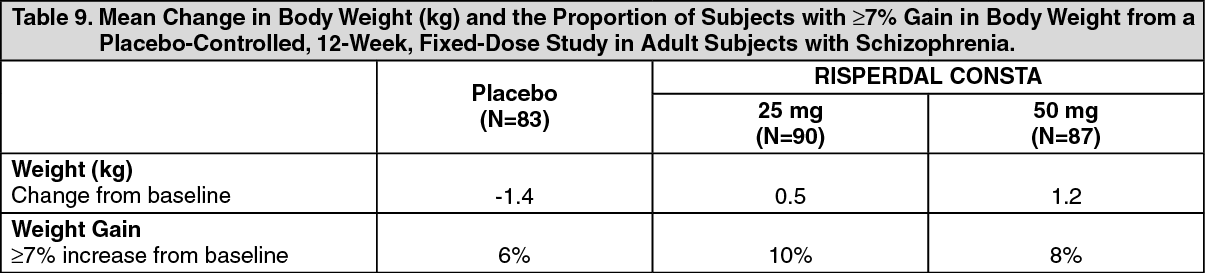 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image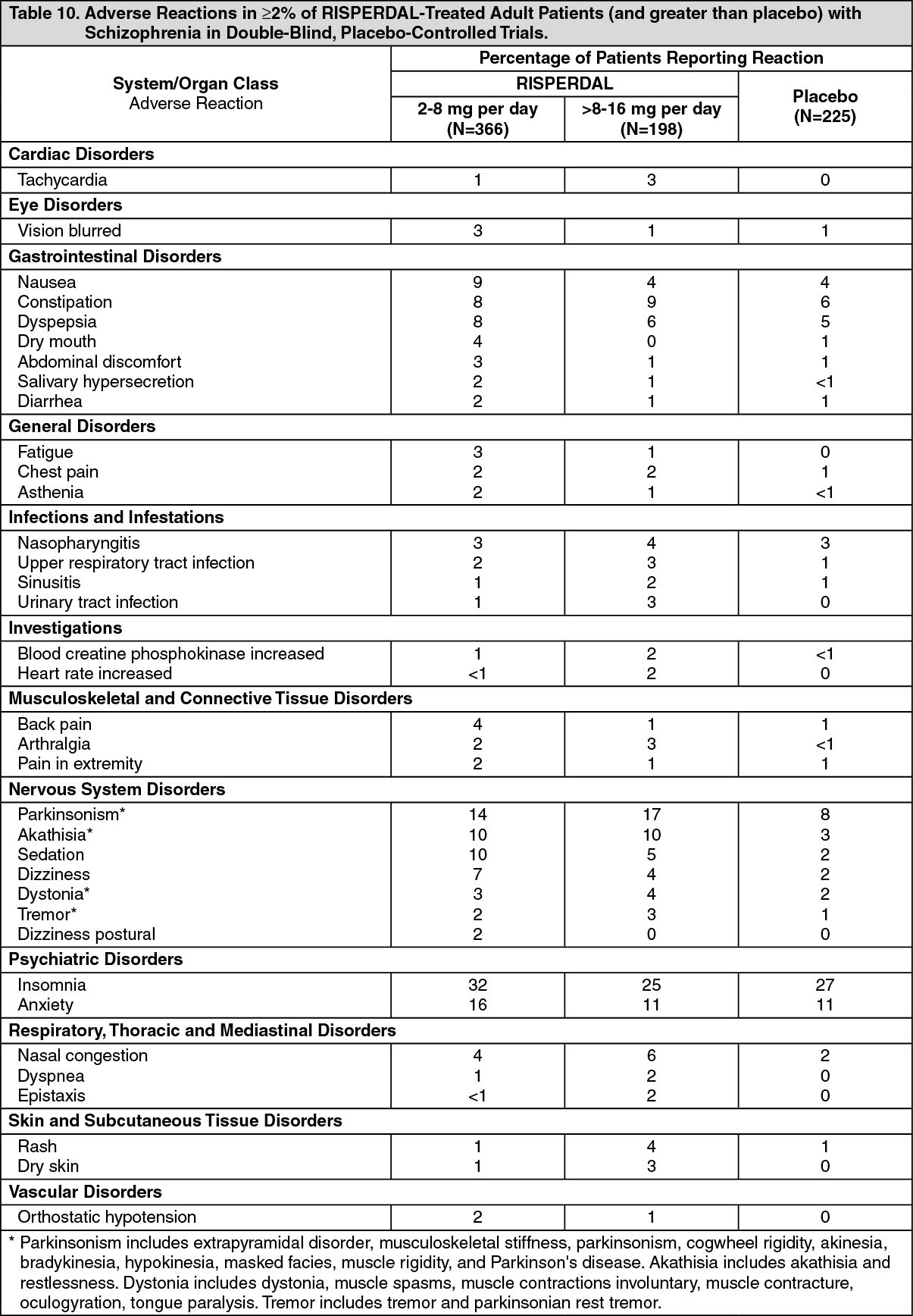 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image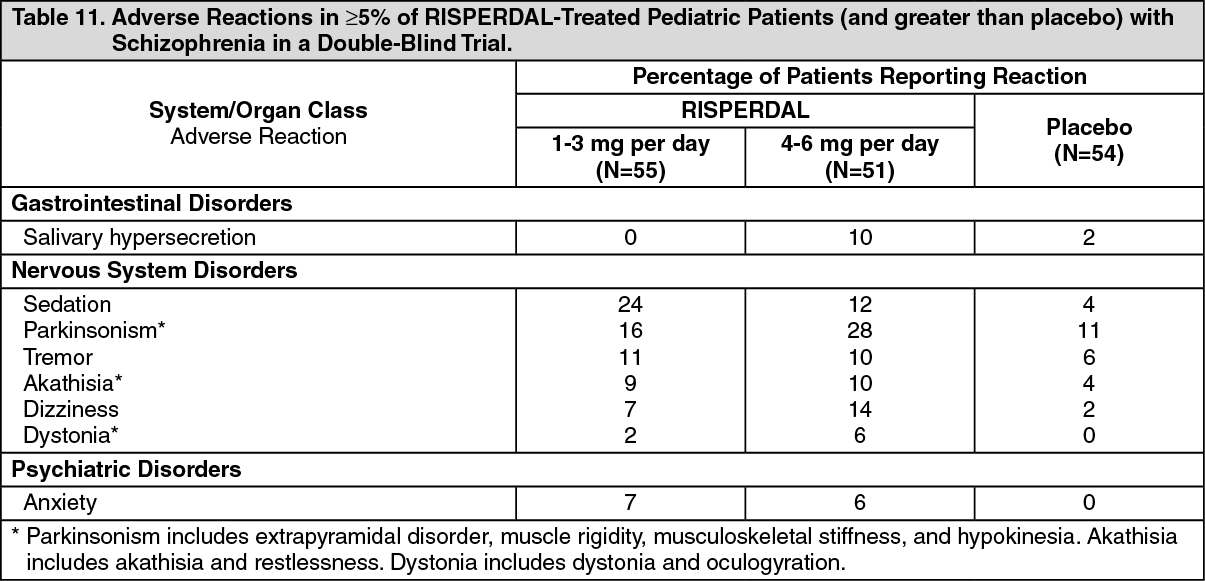 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image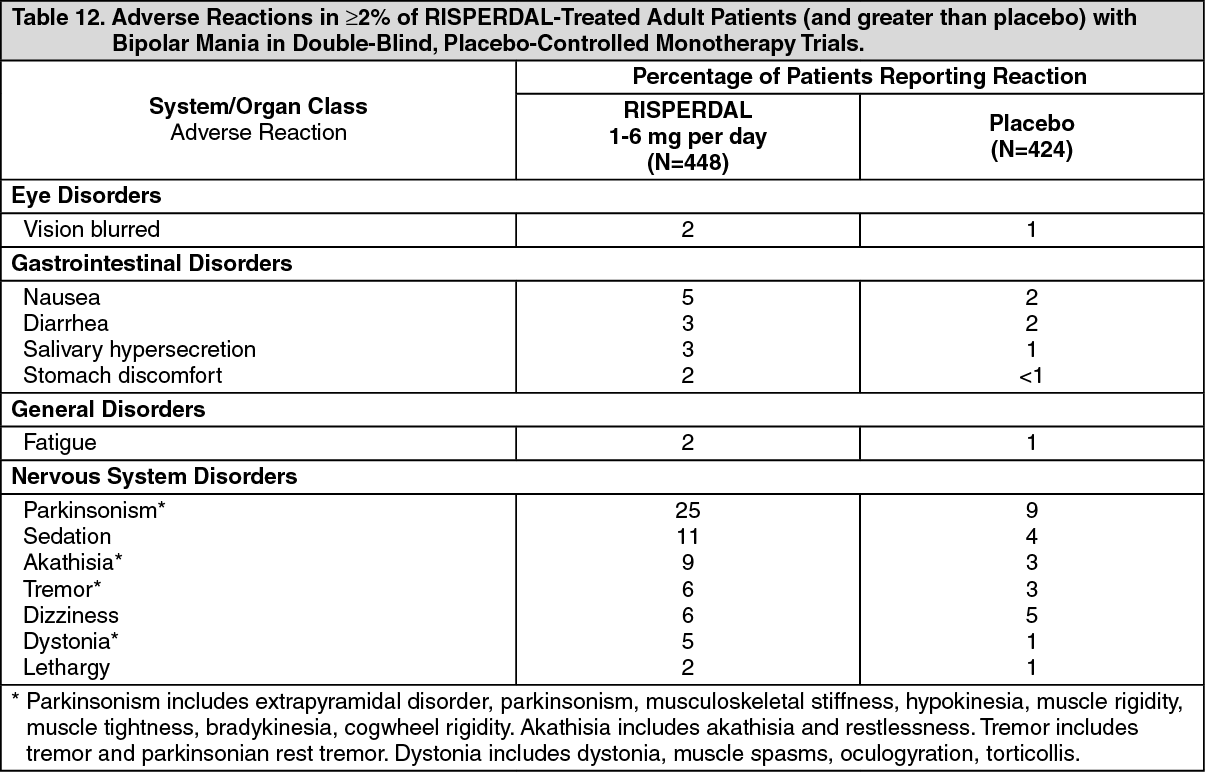 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image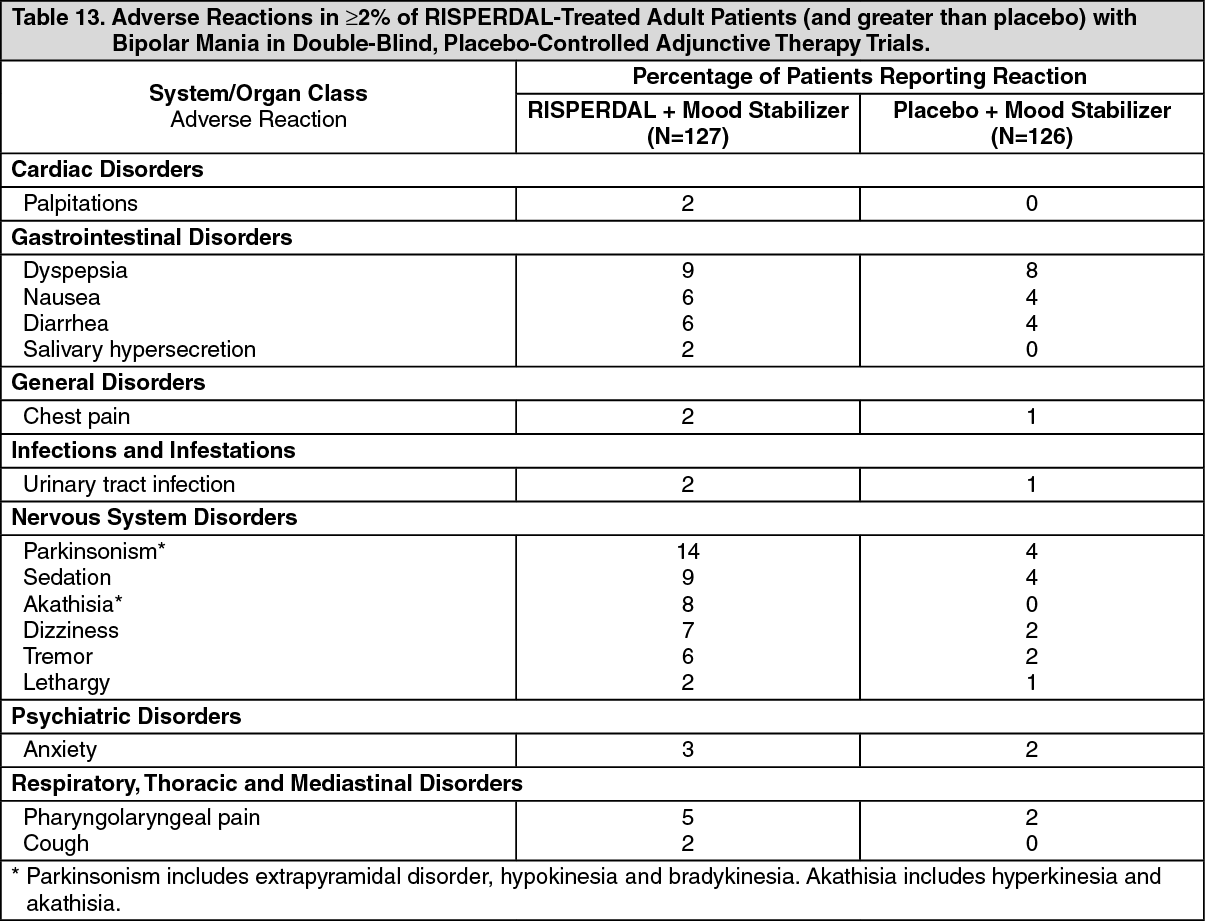 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image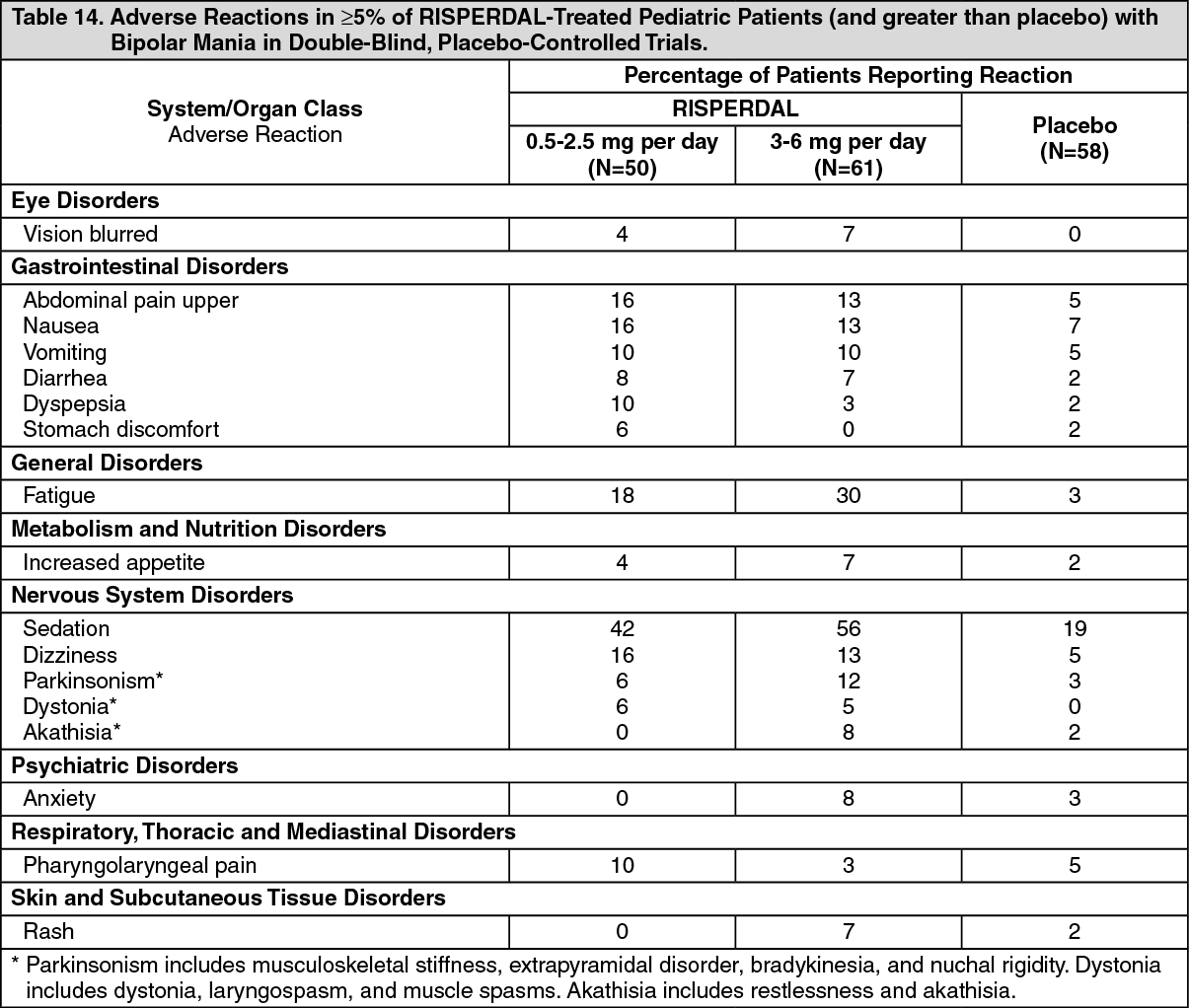 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image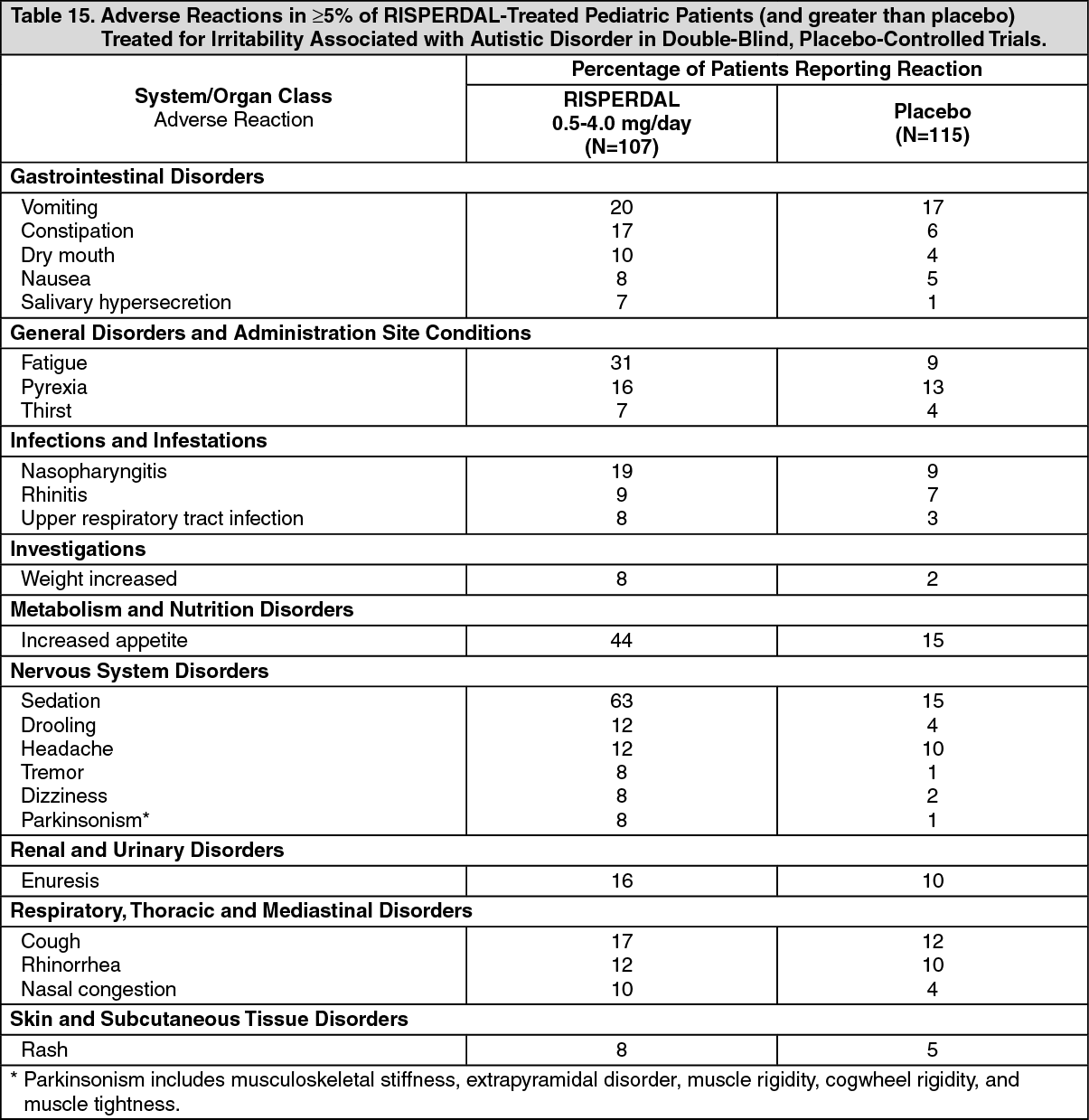 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image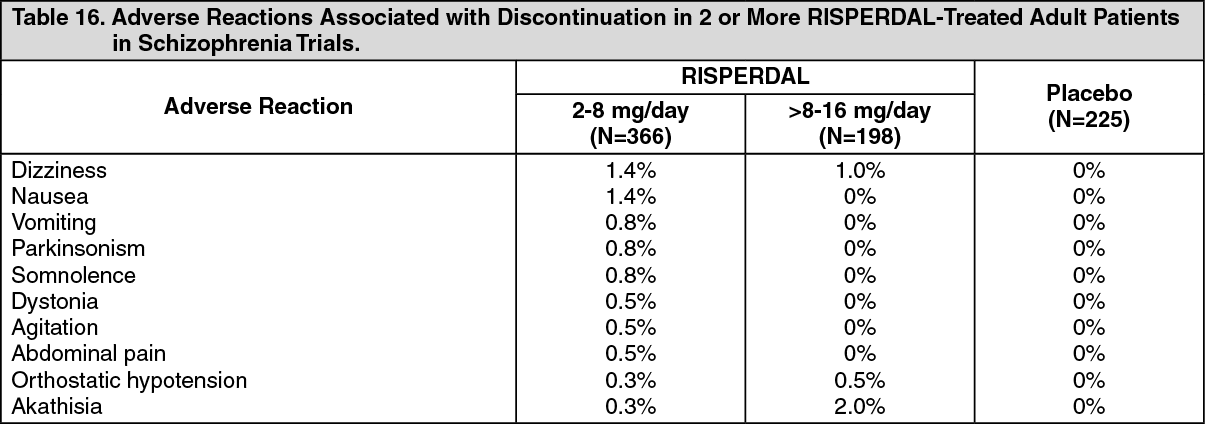 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image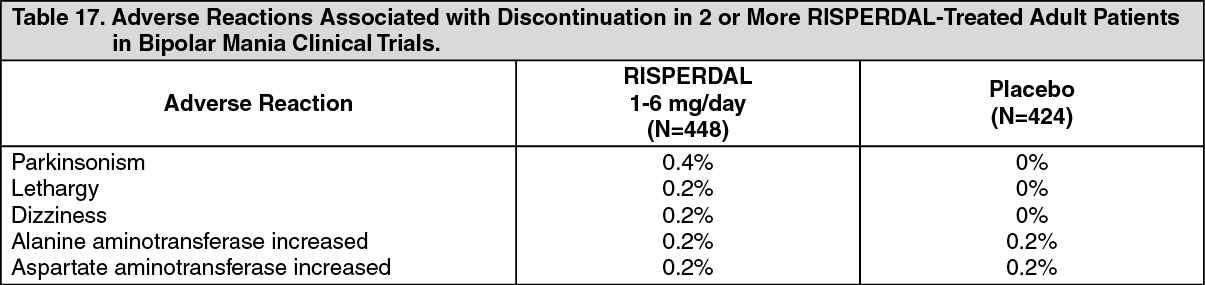 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image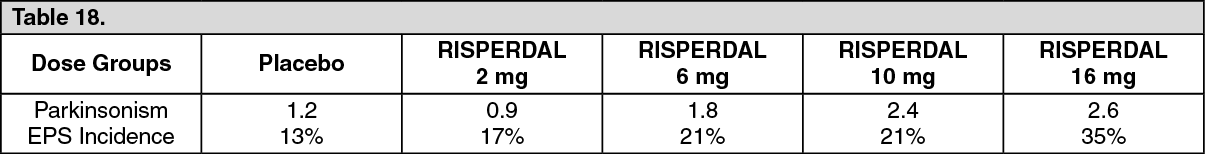 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image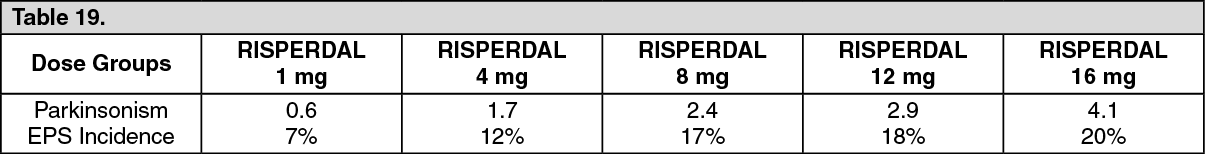 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image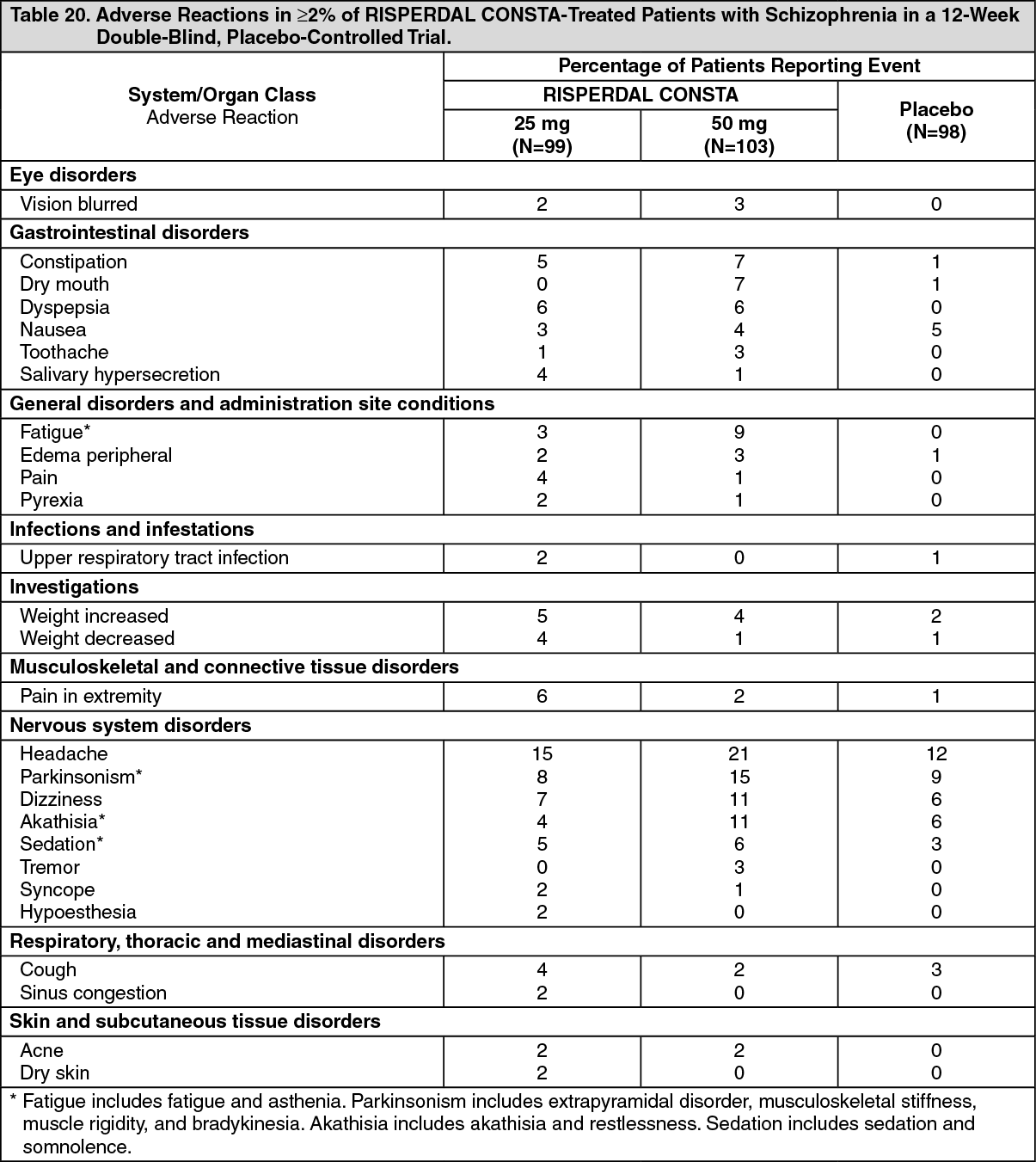 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image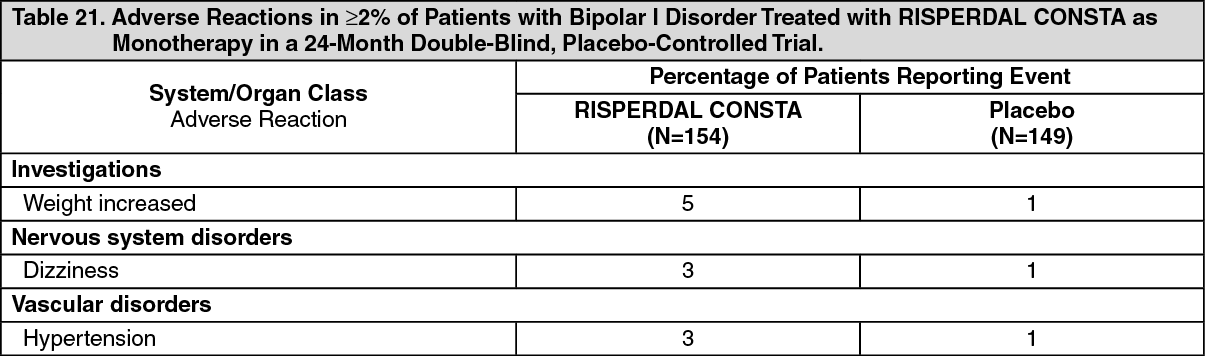 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image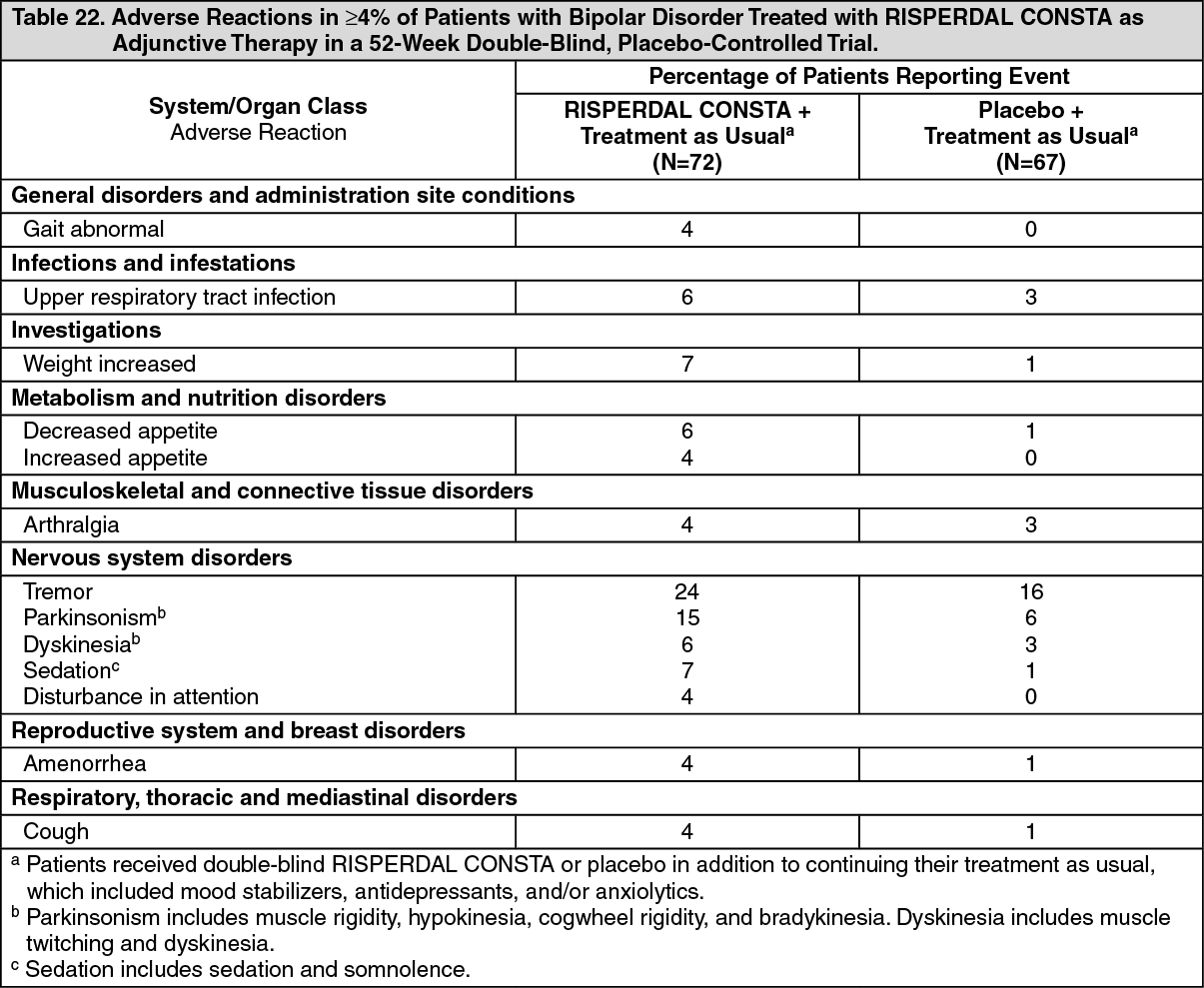 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image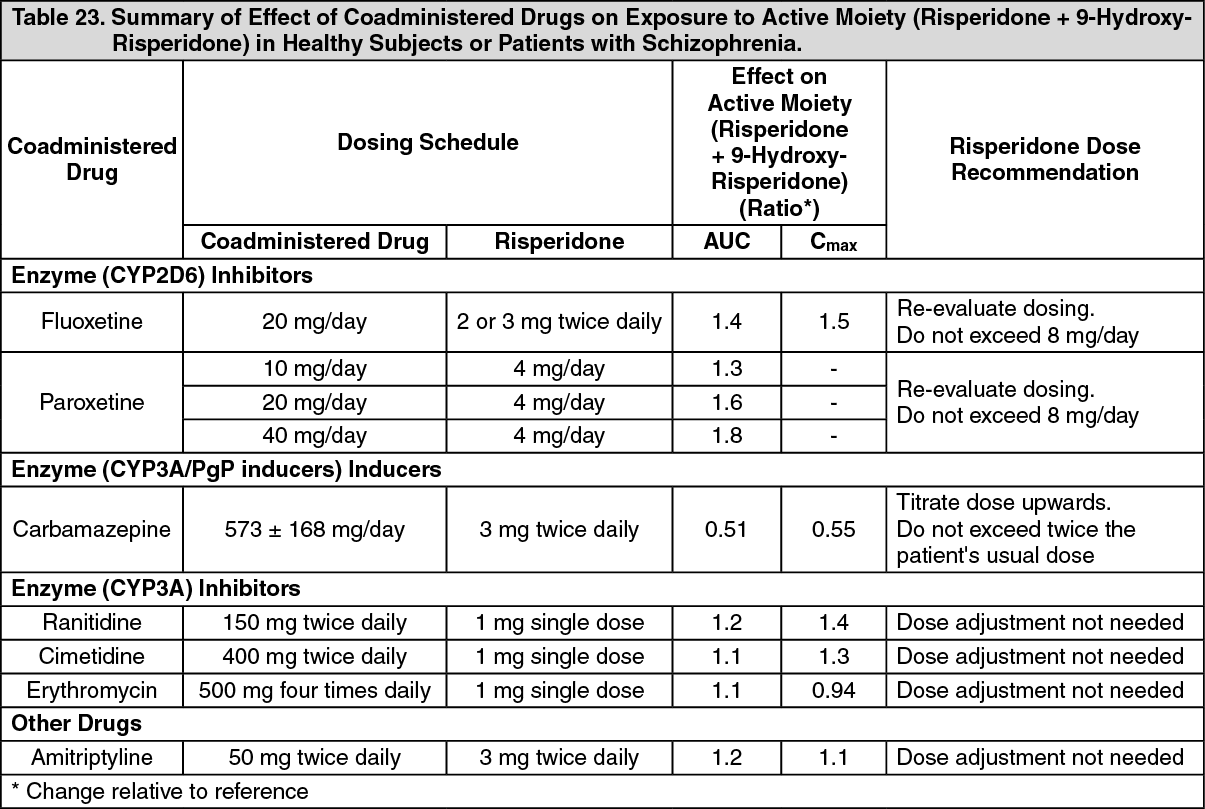 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image




 Sign Out
Sign Out